Loote Ultrahelikeskus начала предлагать новый уникальный неинвазивный пренатальный скрининговый тест GeneSafe Complete.
Тест GeneSafe Complete – это пренатальный неинвазивный скрининговый тест. Посредством данного теста возможно выявить около 29 генных мутаций у плода, способных вызывать 49 различных генетических заболеваний. При помощи ультразвуковой диагностики эти отклонения обнаруживаются далеко не всегда и нередко проявляются лишь на поздних стадиях беременности или даже в первые годы жизни ребенка.
Что представляет собой тест GeneSafe Complete?
Для проведения теста GeneSafe Complete используется образец материнской крови, в котором имеется ДНК и матери и плода (генетический материал). ДНК плода, подлежащая тестированию, поступает из плаценты. Тест GeneSafe Complete выявляет в крови матери ДНК плода, и далее на основе этого оценивается, имеется ли у плода искомое генетическое заболевание или нет.
Определить пол еще не родившегося ребенка посредством данного теста невозможно.
Для проведения теста GeneSafe Complete также необходимо, чтобы биологический отец ребенка сдал анализ слизистой оболочки полости рта.
Тест GeneSafe Complete — это неинвазивный пренатальный скрининговый тест. Для Вас и Вашего ребенка прохождение данного теста не представляет абсолютно никакой опасности: у Вас берется образец крови из вены, а изо рта отца ребенка, со слизистой оболочки щеки ватным тампоном снимается соскоб, содержащий образец ДНК. Полученный генетический материал отправляется экспресс-курьером в итальянскую лабораторию Eurofins Genoma, находящуюся в Риме. Лаборатория Eurofins Genoma известна 20-летним опытом пренатальных генетических исследований. Ежегодно в этой лаборатории проводится более 200 000 генетических тестов.
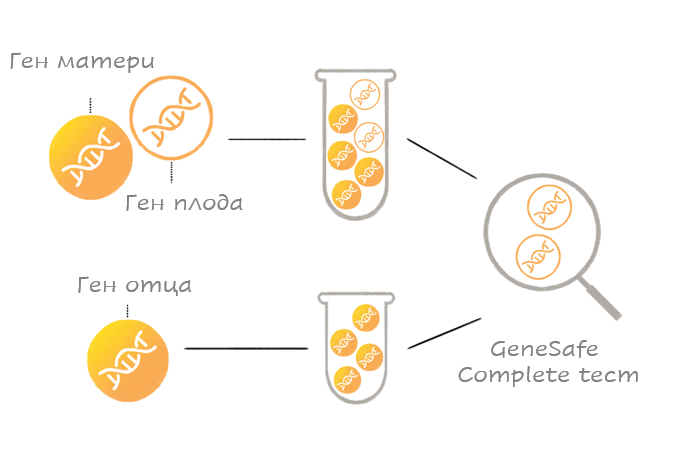
О чем мне скажет пренатальный тест GeneSafe Complete?
Пренатальный тест GeneSafe Complete позволяет выявлять моногенные заболевания, т.е. заболевания, наследуемые по законам Менделя. Моногенные заболевания вызываются мутацией одного или обоих генов (аллелей) пары генов.
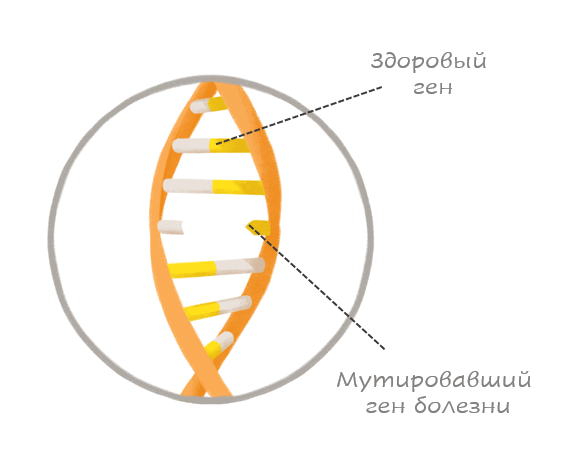
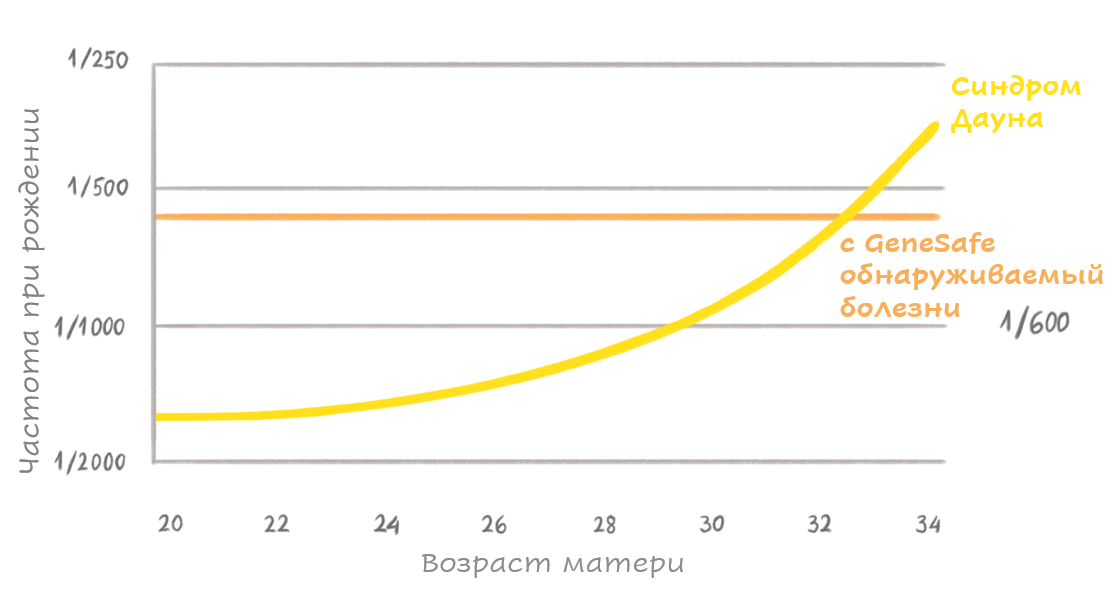
В большинстве случаев подобные заболевания вызываются исключительно генной мутацией, и окружающая среда на их развитие никоим образом не влияет. Наследуемые таким путем заболевания также еще называются менделевскими заболеваниями, поскольку закономерность их наследования была в свое время установлена Грегором Менделем. Практически все они относятся к редким заболеваниям, то есть встречаются реже, чем у одного плода из двух тысяч.
Поскольку тест GeneSafe Complete выявляет 49 моногенных заболеваний, комбинированная частота встречающихся заболеваний составляет приблизительно 1 : 600, то есть один случай на шестьсот плодов; это выше, чем заболеваемость синдромом Дауна.
Проявление моногенных заболеваний происходит по принципу доминантного (преобладающего) или рецессивного (скрытого) наследования. Если ген, вызывающий заболевание, расположен в аутосоме (хромосомы 1-22), то речь идет об аутосомном наследовании и болезнь проявляется одинаково вне зависимости от пола.
Тест GeneSafe Complete — это скрининговый тест, который позволяет выявлять как аутосомно-рецессивные заболевания, унаследованные от родителей, так и заболевания, вызванные новыми мутациями (de novo), возникшими после оплодотворения яйцеклетки.
В случае новых мутаций ген заболевания (например, синдром Ретта или Нунан) не наследуется ни от одного из родителей, а возникает во время формирования сперматозоида или яйцеклетки. Новые мутации особенно заметны именно в случае аутосомно-доминантных заболеваниях, потому что они проявляются сразу – у здоровых родителей, к их удивлению, рождается больной ребенок. Родившийся ребенок в дальнейшем способен теоретически в 50% случаев передать болезнь своему потомству.
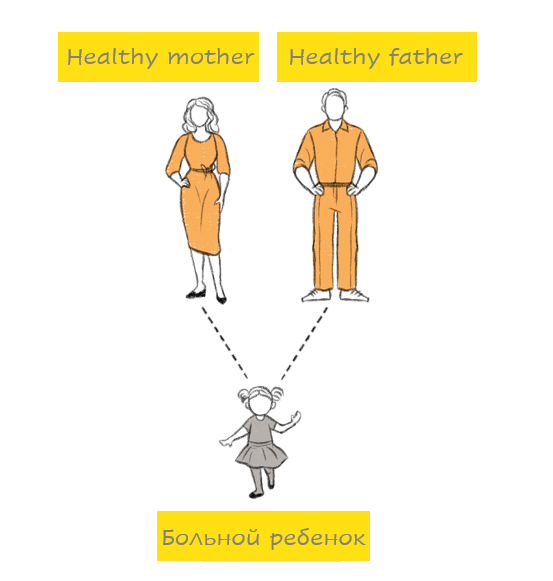
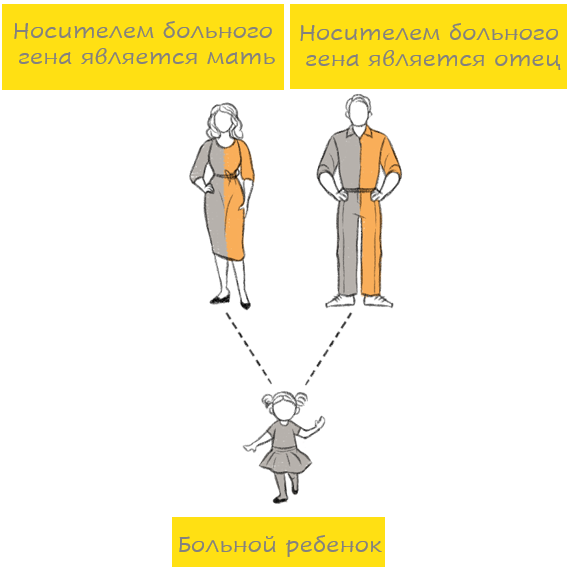
Аутосомно-рецессивная мутация наследуется от обоих родителей (например, муковисцидоз, врожденная нейросенсорная глухота). Это означает следующее: для того, чтобы у ребенка проявилось генетическое заболевание, необходимо изменение в обоих генах одной генной пары. Если ребенок наследует от одного из родителей один измененный ген, а другой ген при этом остается нормальным, то в большинстве случаев нормальный ген компенсирует измененный ген. В этом случае ребенок является носителем мутации, хотя сам при этом здоров. Если же родители оба являются носителями одной и той же генной мутации, то они передают своему ребенку либо нормальный ген, либо измененный. Передача гена происходит случайно, без какой бы то ни было закономерности. Таким образом, каждый ребенок, родители которого оба являются носителями одного и того же измененного гена, имеет риск 1 из 4 (25%) наследовать мутировавший ген как от отца, так и от матери, то есть родиться с наследственным заболеванием. Однако при этом существует также вероятность 3 из 4 (75%), что у ребенка не будет наследственного заболевания.
Поскольку при аутосомно-рецессивных заболеваниях гены, вызывающие заболевание, расположены в аутосомных хромосомах (хромосомы 1-22), заболевание проявляется как у мальчиков, так и у девочек. Подобный риск существует при каждой беременности.
Насколько надежен тест GeneSafe Complete?
Данный скрининговый тест может обнаружить более 99% из 29 генных мутаций. Необходимость повторения теста из-за неясного результата составляет менее 1%.
Каковы альтернативы тесту GeneSafe Complete?
Тест GeneSafe Complete – это скрининговый тест на выявление моногенных заболеваний. При помощи теста GeneSafe Complete невозможно обнаружить хромосомные нарушения, микроделеции или установить пол еще не родившегося ребенка. С помощью теста GeneSafe Complete также невозможно оценить анатомические структуры плода или возможные осложнения, которые могут сопутствовать беременности – такие как преэклампсия и задержка роста плода.
Если Вы хотите также получить информацию о хромосомных патологиях плода, микроделециях, развитии структур органов плода и осложнениях во время беременности, возможно прохождение теста SMART, который в дополнение к тесту GeneSafe Complete включает в себя еще два теста: тест KaryoPlus и тест OSCAR.
Если вы хотите узнать около 2000 различных генетических заболеваний, унаследованных от родителей, в дополнение к генным заболеваниям, исследуемым с помощью теста GeneSafe Complete, вам может потребоваться тест WISE, который в дополнение включает тест KaryoPlus, тест OSCAR и тест GeneScreen Complete. к тесту GeneSafe Complete.
Для женщин, у которых ультразвуковое исследование выявляет подозрение на порок развития плода, предусмотрены инвазивные диагностические тесты – такие как биопсия хориона или анализ околоплодных вод. Посредством данных тестов выявляется более 99% всех хромосомных аномалий, включая редкие генетические аномалии, которые не обнаруживаются с помощью теста GeneSafe Complete и других скрининговых тестов. При проведении диагностических исследований необходимо учитывать, что в 0,1% случаев они могут стать причиной выкидыша независимо от того, была ли у плода хромосомная патология или нет.
Когда я узнаю результаты своего теста?
Вы узнаете результаты теста через 21 день после сдачи анализа крови.
Каких результатов я могу ожидать от теста GeneSafe Complete?
Ответ в отношении теста Vistara, получаемый из лаборатории Natera, включает один из следующих вариантов.
- ОТРИЦАТЕЛЬНЫЙ РЕЗУЛЬТАТ СКРИНИНГОВОГО ТЕСТА
Отрицательный результат скринингового теста означает, что у вашего плода не обнаружено патогенных или вероятно патогенных мутаций в 29 генах, исследованных в рамках теста GeneSafe Complete.
- ПОЛОЖИТЕЛЬНЫЙ РЕЗУЛЬТАТ СКРИНИНГОВОГО ТЕСТА
Положительный результат скрининга означает, что у Вашего плода очень высока вероятность наличия одного из моногенных заболеваний, выявляемых с помощью теста GeneSafe Complete, но это не на 100 % так. В случае такого ответа необходимо подтвердить или исключить наличие генетического заболевания. Для этого требуется диагностический тест – такой как биопсия хориона или анализ околоплодных вод, или же исследование венозной крови новорожденного, взятой после рождения.
Кому можно проводить этот тест и когда?
Тест Vistara предназначен для женщин всех возрастов и любого этнического происхождения, беременность которых длится не менее 10 полных недель (10 недель + 0 дней). Если вы хотите узнать срок своей беременности, воспользуйтесь нашим калькулятором беременности.
Тест GeneSafe Complete также может быть предложен:
- при ЭКО беременности;
- при беременности двойней;
- при беременности двойней, если один плод погиб;
- при одноплодной и двуплодной беременности, если для искусственного оплодотворения использовались донорские яйцеклетки или донорская сперма;
- если биологический отец страдает генетической патологией, которую можно исследовать с помощью GeneSafe Complete.
Тест GeneSafe Complete нельзя предлагать:
- в случае тройни;
- если матери крови в течение последнего месяца переливалась кровь;
- если мать страдает одной из генетических патологий, обнаруживаемых с помощью теста GeneSafe Complete.
Кому будет особенно полезен тест GeneSafe Complete?
Частота моногенных заболеваний не зависит от возраста женщины, но они чаще возникают, если биологический отец будущего ребенка пожилой. Мы рекомендуем пройти тест GeneSafe Complete, если отец ребенка старше 40 лет.
Особенно полезен тест женщинам:
- биологический отец ребенка которых старше 40 лет;
- которые хотят получить о своем ребенке как можно больше информации;
- по результатам УЗИ которых появилось подозрение на моногенное заболевание плода, но женщина не желает проходить инвазивное исследование;
- у ребенка которых, судя по анамнезу, повышен риск развития кистозного фиброза или генетической глухоты;
- у плода которых по результатам УЗИ в третьем триместре обнаружены очень короткие трубные кости или череп плода очень мал или имеет очень необычную форму (подозрение на преждевременное закрытие черепных швов).
Большинство женщин, прошедших тест Vistara, узнают, что у их ребенка низкий риск возникновения протестированных моногенных заболеваний – что может быть очень ободряющей новостью.
Какие моногенные заболевания можно выявить, пройдя тест GeneSafe Complete?
-
Синдромальные краниосиностозы
Краниосиностоз вызывается преждевременным окостенением черепных швов, что приводит к развитию характерной формы головы и черт лица. В некоторых случаях краниосиностоз может вызывать различные неврологические нарушения и проблемы с питанием. Кроме того, заболевание может повысить внутричерепное давление и вызвать нарушения зрения. Краниосиностоз может быть несиндромальным или же сопровождаться другими генетическими синдромами. Примерно в 85% случаи краниосиностоза являются несиндромальными. Краниосиностоз встречается с частотой приблизительно 1:2000–2500 живорождений.
- Синдром Антли-Бикслера (FGFR2).
Наблюдается преждевременное окостенение черепа, искривленные трубчатые кости и пальцы, затвердение суставов пальцев. Может сопровождаться пороками развития сердца, отсутствием анального отверстия, ноздрей и влагалища. - Синдром Апера (FGFR2).
Наблюдается преждевременное окостенение черепа, сращение пальцев рук и патология прикуса. - Синдром Крузона (FGFR2, FGFR3).
Наблюдается преждевременное окостенение черепа, в некоторых случаях приводит к потере слуха и проблемам прикуса. - Синдром Джексона-Вейсса (FGFR2).
Наблюдается преждевременное окостенение черепа и отклонения в развитии ног. - Синдром Пфейффера, тип 1, 2, 3 (FGFR2).
Наблюдается преждевременное окостенение черепа, может наблюдаться потеря слуха, умственная отсталость, отклонения в развитии рук и ног. В более тяжелых случаях новорожденный может погибнуть после родов.
- Синдром Антли-Бикслера (FGFR2).
-
Неврологические состояния
В случае неврологических состояний наблюдаются проблемы в развитии центральной нервной системы, что приводит к задержке психомоторного развития детей и часто к нарушению интеллекта и умственной отсталости.
- Синдром Ретта (MECP2).
В большинстве случаев наблюдается у девочек. В возрасте от 6 до 18 месяцев наблюдается быстрое снижение развития речевых и моторных навыков. Часто встречаются аутизм и судорожный синдром.
- Синдром Ретта (MECP2).
-
Нарушения спектра Нунан
Синдр Нунан – это аутосомно-доминантное наследственное заболевание, для которого характерны низкий рост, врожденный порок сердца и задержка психического развития различной степени тяжести. Пациенты с синдромом Нунан имеют характерную внешность: короткая шея, кожные складки на шее, низко посаженные уши, гипертелоризм. Кроме того, может наблюдаться дисплазия лимфатической системы, которая приводит к кистозной гигроме и увеличению шейной складки. Частота возникновения синдрома Нунан составляет ~1:1000–2500.
- Кардиофациокутанеальный синдром (BRAF, MAP2K1, MAP2K2).
Характерны низкий рост, измененные черты лица, общий ихтиоз, пороки развития сердца. Может вызвать задержку развития и умственную отсталость. - Синдром Нунан 1, 3, 4, 5, 6, 8 (PTPN11, RAF1, RIT1, KRAS, NRAS, SOS2, SHOC2, BRAF, MAP2K1, CBL).
Характерны специфические черты лица, типичные для данного заболевания, низкий рост, патология скелета, пороги развития сердца, нарушение свертываемости крови, нарушение опускания яичек. В некоторых случаях – легкая умственная отсталость. - Синдром LEOPARD 1,2 (PTPN11, RAF1).
- Схож с синдромом Нунан, с выраженными коричневыми пятнами на коже (лентигиноз), низкий рост, пороги развития сердца, кровотечения, возможна умственная отсталость различной степени тяжести.
- Ювенильный миеломоноцитарный лейкоз (PTPN11).
Наблюдается агрессивный лейкоз с быстрым течением. 5-летнее выживание в 50 % случаев.
- Кардиофациокутанеальный синдром (BRAF, MAP2K1, MAP2K2).
-
Нарушения, связанные с костной системой
Существует более 200 врожденных нарушений роста костей. Наиболее часто встречающимися нарушениями, связанными с костной системой, являются остеохондродисплазии, которые затрагивают как длинные трубчатые кости и позвоночник, так и хрящевые компоненты костей. Часто эти заболевания сопровождаются задержкой роста ребенка, деформацией костей и позвоночника. При этом умственное развитие детей часто соответствует их возрасту. При более тяжелых формах заболеваний дети погибают внутриутробно или сразу после родов вследствие дыхательной недостаточности.
- Ахондроплазия (FGFR3).
Наблюдается нарушение роста скелета, для которого характерны выраженная карликовость, в частности укороченная плечевая и бедренная кость, непропорционально большая голова и сужение позвоночного канала. - Синдром CATSHL (FGFR3).
Характерны высокий рост, боковое искривление позвоночника, потеря слуха, затвердение суставов пальцев. Может сопровождаться умственной отсталостью. - Несовершенный остеогенез, тип I, II, III, IV (COL1A1, COL1A2).
Крайне ломкие кости, часто без видимой причины. В более тяжелых случаях новорожденные погибают в результате дыхательной недостаточности. - Синдром Элерса-Данло, классический, тип VIIA; форма сердечного клапана, тип VIIB (COL1A1, COL1A2.
Характерны специфические черты лица, типичные для данного заболевания, повреждения соединительной ткани и гипермобильные суставы. Может сопровождаться опасным для жизни осложнением, например аневризмой аорты. - Гипохондроплазия (FGFR3).
Нарушение роста скелета, сопровождающееся низким ростом и сужением позвоночного канала. Может сопровождаться судорожным синдромом со вторичной задержкой развития. - Танатотрофная дисплазия, типы I, II (FGFR3).
Тяжелая задержка роста и патология скелета. Ребенок часто погибает в утробе матери или в постнатальный период из-за дыхательной недостаточности.
- Ахондроплазия (FGFR3).
-
Врожденные синдромы
Под синдромами подразумевается совокупность симптомов, характерных для определенного заболевания или их комбинаций. Зачастую синдромы затрагивают несколько систем органов одновременно и связаны с умственной недоразвитостью.
- Синдром Алажилля (JAG1).
Наблюдаются пороки развития сердца и проблемы с печенью. Может сопровождаться проблемами роста и отклонениями позвоночника. - Синдром CHARGE (CHD7).
Наблюдается закрытие носовых ходов, патология сетчатки глаза, пороки развития сердца, отклонения в половых органах, аномальные уши, потеря слуха, задержка роста и развития, а также заячья губа и/или волчья пасть. - Синдром Корнелии де Ланге 1, 2, 3, 4, 5 (NIPBL, SMC1A, SMC3, RAD21, HDAC8).
Характерны специфические черты лица, типичные для данного заболевания, задержка роста и развития, а также умственная отсталость различной степени тяжести. - Синдром Мюнке (FGFR3).
Наблюдается преждевременное окостенение черепа, может наблюдаться потеря слуха, психомоторная задержка развития, а также заячья губа и/или волчья пасть. - Синдром Сотоса 1 (NSD1).
Быстрый рост, наблюдается психомоторная задержка развития, когнитивная и умственная недоразвитость. Могут наблюдаться расстройства аутистического спектра. - Синдром Боринга-Опица (мутация гена ASXL1). Отмечается задержка роста; характерные признаки заболевания – маленькая голова и дисморфизм лица с выпученными глазами. Нередко наблюдаются контрактуры локтевых и лучезапястных суставов (ограниченность сгибания и разгибания), выраженные нарушения питания; впоследствии – трудности в обучении.
- Синдром Шинцеля-Гидеона (мутация гена SETBP1).
Характерными особенностями являются специфические черты лица, чрезмерный рост волос и судорожный синдром, а также аномалии скелета: короткие верхние и нижние конечности. Кроме того, отмечается глубокая прогрессирующая умственная отсталость. - Голопрозэнцефалия (мутация гена SIX3).
Характеризуется тяжелым нарушением развития: передний мозг остается неразделенным на полушария, чему нередко сопутствуют деформации лица. В тяжелых случаях прогноз крайне неблагоприятный: многие дети живут не дольше шести месяцев. В более легких случаях больные могут дожить до совершеннолетия, но при этом нередко сохраняются различные неврологические дефекты и недостатки развития
- Синдром Алажилля (JAG1).
-
Болезни, унаследованные от родителей
При наследственных аутосомно-рецессивных заболеваниях ребенку от каждого из родителей передается больной ген, т. е. эти заболевания наследуются ребенком от родителей.
- Кистозный фиброз (мутация гена CFTR)
Самое распространенное в Эстонии генетическое заболевание, сокращающее срок жизни. Встречается с частотой 1 случай на 4500 живорождений. Это хроническое, прогрессирующее заболевание, способное поражать все органы и системы. В классических случаях наблюдается прогрессирующее заболевание легких, которое как правило сопровождается одышкой, сахарным диабетом и повышенным содержанием соли в поту. Болезнь может сопровождаться задержкой роста из-за нарушений питания, диареей, печеночной недостаточностью и бесплодием. Клиническая картина и тяжесть заболевания могут варьировать от множественного поражения органов до легкого поражения одного органа. Симптомы обычно проявляются уже в первые годы жизни ребенка. - Аутосомно-рецессивная глухота типа 1А, 1В (обусловлена мутациями генов CX26, GJB2, CX30, GJB6)
Врожденная несиндромальная генетическая сенсоневральная тугоухость различной степени тяжести на 70% обусловлена именно такими генными мутациями. Сенсоневральная потеря слуха представляет собой хроническое и в настоящее время неизлечимое заболевания. - Бета-талассемия (HBB)
При бета-талассемии синтезируется мало гемоглобина нормальной структуры, в результате чего у ребенка развивается анемия. Заболевание проявляется в течение первых двух лет жизни. У ребенка отмечается бледность, замедленная прибавка в весе или росте, диарея и рецидивирующая лихорадка. Из-за увеличения печени отмечается также вздутие живота. При лечении тяжелой формы талассемии проводят трансплантацию гемопоэтических стволовых клеток. - Серповидноклеточная анемия (HBB)
При серповидноклеточной анемии в эритроцитах присутствует аномальный гемоглобин. Это в свою очередь приводит к образованию серповидных эритроцитов с нарушенными функциями и укороченным сроком жизни. Чаще встречается у чернокожих детей в первые четыре месяца жизни. Когда серповидные эритроциты начинают массово разрушаться или костный мозг не в состоянии производить достаточного количества новых эритроцитов, то серповидноклеточная анемия представляет опасность для жизни.
- Кистозный фиброз (мутация гена CFTR)
Большинство женщин, прошедших тест GeneSafe Complete, обнаруживают, что у их ребенка имеется минимальный риск развития моногенного заболевания, на которое женщина проверялась, – это может очень ободрить и обнадежить.